Article Text

Abstract
Introduction The Bioceptive suction cervical retractor (SCR) is a novel device that can replace the standard single-tooth tenaculum to place traction on the cervix. A feasibility trial was conducted on the device for intrauterine device (IUD) placement.
Methods Our three-stage feasibility process began with Stage 1, where the device was tested on in-vitro and ex-vivo samples. In Stage 2, 10 women received their IUD using the device. In Stage 3, a feasibility trial, we randomly assigned 25 consenting women to receive their IUD using either the Bioceptive SCR or the standard single-tooth tenaculum. In Stages 2 and 3, we collected pain scores using an electronically adapted 100-point visual analogue scale (VAS) at eight timepoints during and after the insertion procedure, as well as satisfaction and acceptability measures. The primary outcome was the pain score after attaching the SCR or tenaculum (VAS 3). Wilcoxon rank sum tests compared pain scores between devices.
Results In Stage 2, pain scores with the SCR were lower than historical controls with the single-tooth tenaculum. In Stage 3, the median VAS 3 pain scores were 31 and 57 for the intervention and control groups, respectively. The differences in pain scores were not statistically significant but the trend was to lower pain scores with the intervention. Reported patient satisfaction with the SCR device was 80% in Stage 2% and 90% in Stage 3.
Conclusions The Bioceptive SCR has potential as an atraumatic alternative to standard cervical retractor devices for gynaecological procedures. These findings can guide point estimates for future clinical studies.
Trial registration NCT02283463.
- intrauterine devices
- pain
- cervical retractor
- bioceptive
- iud insertion
Statistics from Altmetric.com
Key messages
Improvements in intrauterine device (IUD) insertion and gynaecological procedures are needed to reduce discomfort and pain in these procedures and facilitate ease of procedures.
The Bioceptive suction cervical retractor was designed as an atraumatic alternative to standard cervical retraction devices.
Initial feasibility testing shows that use of the device for IUD insertion appears to be a safe and clinically acceptable for both patients and providers.
Introduction
Intrauterine device (IUD) use is increasing in the United States (US), yet fear of insertion pain remains a barrier to uptake.1–18 A substantial percentage of women receiving IUDs (~11%–17%, depending on parity) report severe pain during insertion.2 4 This includes pain experienced during cervical traction, which can be comparable to pain experienced during the device insertion.6 As a result, reducing insertion pain has been a focal point of many recent studies.6–13 16–18 However, a small minority of tested interventions have demonstrated successful pain reductions.6 14 15
At present, the most common medical device used to place traction on the cervix in the US is the standard single-tooth tenaculum, a two-pronged tool that penetrates opposing points into the cervical stroma. The tenaculum effectively engages the cervix; however, puncture of the cervical tissue is associated with pain and bleeding.9 15 Recent studies comparing pain scores during IUD insertions with the single-tooth tenaculum and less traumatic instruments have yielded variable results.9 16 17
The Bioceptive suction cervical retractor (SCR) was developed to streamline cervical traction and transcervical gynaecological procedures, while providing a less painful, atraumatic alternative to the single-toothed tenaculum (figure 1). Since the SCR device retracts the cervix via suction, rather than puncture of cervical tissue, procedures using it are theorised to be atraumatic to the cervix and potentially less painful. This article reports three stages in the feasibility process of implementing the SCR device for use during IUD insertion procedures, assessing acceptability, implementation, practicality, and limited efficacy testing to determine whether to move forward with further testing for the SCR.19 We assess patient-reported pain and satisfaction during and after IUD insertion for the SCR device. The trial adheres to the CONSORT checklist for pilot or feasibility trials (see Appendix A in Eldridge et al).20

Engagement of cervix and dilation with the Bioceptive suction cervical retractor (SCR). (A), (B) Provider aligns suction port with external cervical os. (C) Suction gently pulls cervix into suction port, simultaneously dilating the initial segment of the cervical canal.
Methods
Overall approach
The reusable Bioceptive SCR is designed to be used in any procedure where an instrument <6 mm in diameter passes through the internal cervical os. The plastic suction cup is discarded after a single use. In this study, we only evaluate its use in IUD insertions. We assessed the feasibility of the SCR device in three separate stages: (1) device concept testing, (2) initial acceptability testing and (3) a randomised, limited-efficacy trial. The study used a single, experienced provider to facilitate standardised exposure and testing feasibility before having less proficient providers use the device. Both Stages (2 and 3) involving human subjects were Institutional Review Board approved and and registered (ClinicalTrials.gov: NCT02283463). The protocol for Stages 2 and 3 are available in online supplementary appendix A.
Supplementary file 1
Stage 1: Concept testing
Prior to clinic testing, extensive testing of the SCR device was conducted on synthetic uterine models, two human cadavers, and 16 ex-vivo hysterectomy samples. These tests identifed appropriate application techniques, force testing with SCR attachment, and demonstrated effective, durable attachment to the cervix. These efforts also informed modifications to optimise attachment strength, minimise surface area and improve visualisation.
Stage 2: Initial acceptability testing
Preliminary device acceptability was tested in ten consenting women undergoing copper IUD insertion at a single clinic in Utah beginning on 3 September 2014. Stage 3 ended on 7 January 2015. This initial testing ensured that the device did not result in insertion pain scores that were outside of the standard of care for IUD insertion. We established an a priori stopping rule should mean or median pain scores on a 100-point visual analogue scale (VAS) at the time of SCR placement (VAS 3 and VAS 4, see below) exceed published values.15 21
Participants
Eligible participants were aged 18–45 years and able to consent in English or Spanish. To recruit participants, trained study staff approached all patients presenting for an IUD insertion about the study. The exclusion criteria included: postmenopausal status, contraindications to IUD insertion on package inserts, cervical abnormalities (eg, cervical polyp, lesion, etc), and narcotic or benzodiazepine use prior to the procedure.22–24 The protocol did not exclude postpartum women, though no participants had ended a pregnancy within 3 months. Participants received a US$50 gift card. Study staff executed the informed consent process for participation in the study and for IUD insertion, as per clinic standard of care.
Procedures
Trained study personnel completed enrollment procedures and administered the Research Electronic Data Capture (REDCap) data collection tool using a tablet computer during the appointment.25 All participants reported demographic data, obstetric and gynaecologic history and any use of pain medications in the 24 hours prior to their procedure.
Participants were informed about the specifics of the SCR device prior to their procedure. With the speculum still in place from normal examination, the SCR device was removed from its packaging and shown to the participant. Attachment of the SCR occurred so that the tip on the suction port entered the cervical os and the outer ring of the port contacted external cervical tissue. While placing gentle forward pressure toward the cervix, the knob on the SCR was turned one full revolution to generate suction. The provider continued to turn the knob until four complete rotations were completed. IUD insertion was accomplished with the IUD manufacturer’s inserter through the SCR.
Outcomes
To assess pain, participants completed an electronicly adapted 100 mm VAS at eight different points during the procedure with anchors of 0 being ‘no pain’ and 100 being the ‘worst pain imaginable’ (table 1).26 Trained study personnel administered the VAS during the insertion process, to ensure appropriate timing for pain level assessments. We placed no limitations on the use of adjunctive measures to facilitate IUD insertion such as cervical dilation, use of ultrasound guidance, or use of local anaesthesia.
The 100-point visual analogue scale (VAS) metrics for pain during intrauterine device (IUD) insertion procedure
We assessed patient post-insertion overall satisfaction using a five-point Likert scale. This question stated: ‘Please rate your level of satisfaction with the procedure’, with response anchors at (1) ‘very dissatisfied’, (3) ‘unsure’ and (5)‘very satisfied’.
Stage 3: Limited efficacy testing
Participants
Finally, we conducted a limited efficacy study using a single-blind randomised design on women receiving either a copper IUD or a levonorgestrel IUD with the same inclusion and exclusion criteria as in Stage 2. Following reassuring pain scores with SCR use in Stage 2, we initiated Stage 3 to assess potential differences in reported pain scores and satisfaction between women blinded and randomised to either IUD insertion with the SCR, or to the standard IUD insertion procedure using the single-tooth tenaculum.
Procedures
The clinician performing IUD insertion was not able to be blinded and the subsequent analysis was not blinded to treatment assignment, though participants were using a drape during the procedure. Stage 3 recruitment and enrollment was the same as Stage 2, but with additional randomisation and patient blinding. In this stage, we did not use cervical anaesthesia prophylactically. A member of the investigative team (JS) generated randomised treatments via REDCap which assigned participants 1:1 to the intervention or control group in blocks of four. The tray was prepared with both standard tenaculum and the Bioceptive SCR, and the research assistant revealed assignments via REDCAP to the provider in the clinical room immediately prior to IUD placement. We sought to enrol 25 participants for this pilot study to inform a point estimate and ranges for pain scores to address power and sample size determinations for the future. Study staff contacted participants the day after IUD insertion by phone, to assess discomfort (VAS #8) or any residual effects of the insertion procedure.
Outcomes and analysis
The primary outcome was the pain score obtained after attaching the suction or single-tooth tenaculum device (VAS 3). We did not pre-specify the primary outcome in the initial protocol as we originally sought to compare pain scores across the IUD insertion process. After trial initiation we choose the primary outcome as the single point to statistically evaluate to most likely differentiate the two approaches. We compared pain scores using Wilcoxon rank sum tests to assess group differences in continuous pain score variables. We also conducted limited comparisons between the groups using Fischer’s exact tests for categorical and demographic variables. All analyses were performed with STATA SE version 14.0 or higher statistical software programme (College Station, TX, USA).
Patient involvement
The Bioceptive SCR device was specifically designed to address patient-reported barriers to IUD uptake due to insertion pain. Participant feedback throughout the various stages of the study, both qualitatively and through satisfaction variables, informed the next study steps.
Results
Table 2 reports demographic information on all participants in Stages 2 and 3 of the study.
Demographic characteristics of women participating in the Bioceptive suction cervical retractor (SCR) study
Stage 1: Concept testing
A main finding from this stage was that the plastic on the original device was translucent and made it difficult to see the cervix during placement. In addition, temperature differences between the room and the vagina created condensation on the device, further obscuring the cervix during insertion. As a result of these findings, the type of plastic on the SCR device was replaced, to improve visibility.
Additionally, with a tilted or obstructed cervix, it was difficult to firmly attach the SCR. During this stage, we also noted that attaching the SCR device resulted in partial dilation of the cervix, which led to improved insertion visibility. The device securely attached to the uterine cervix, creating an atraumatic traction process that did not result in puncture or damage to the cervix.
Stage 2: Initial acceptability testing
Ten women were consented for the acceptability testing stage (demographics in table 2) and table 3 provides an overview of mean/median pain scores at each of the VAS timepoints. VAS 3 and VAS 4 values did not approach established thresholds for study cessation (40 for gel group in Goldthwaite15 and 42 for placebo in Allen21). In two of the procedures, the provider needed more than one attempt to affix the SCR device to the cervix. A single participant had an irregularity of the cervix that made placement difficult, resulting in the need to dilate the cervix in order to insert the IUD. No cervical bleeding occurred during the procedures. Eighty percent of participants reported feeling ‘satisfied’ or ‘very satisfied’ with their experience, immediately following the procedure.
Median patient scores to the 100 mm visual analogue scale (VAS) for pain during intrauterine device (IUD) placement in the Bioceptive suction cervical retractor (SCR) study
Stage 3: Limited efficacy testing RCT
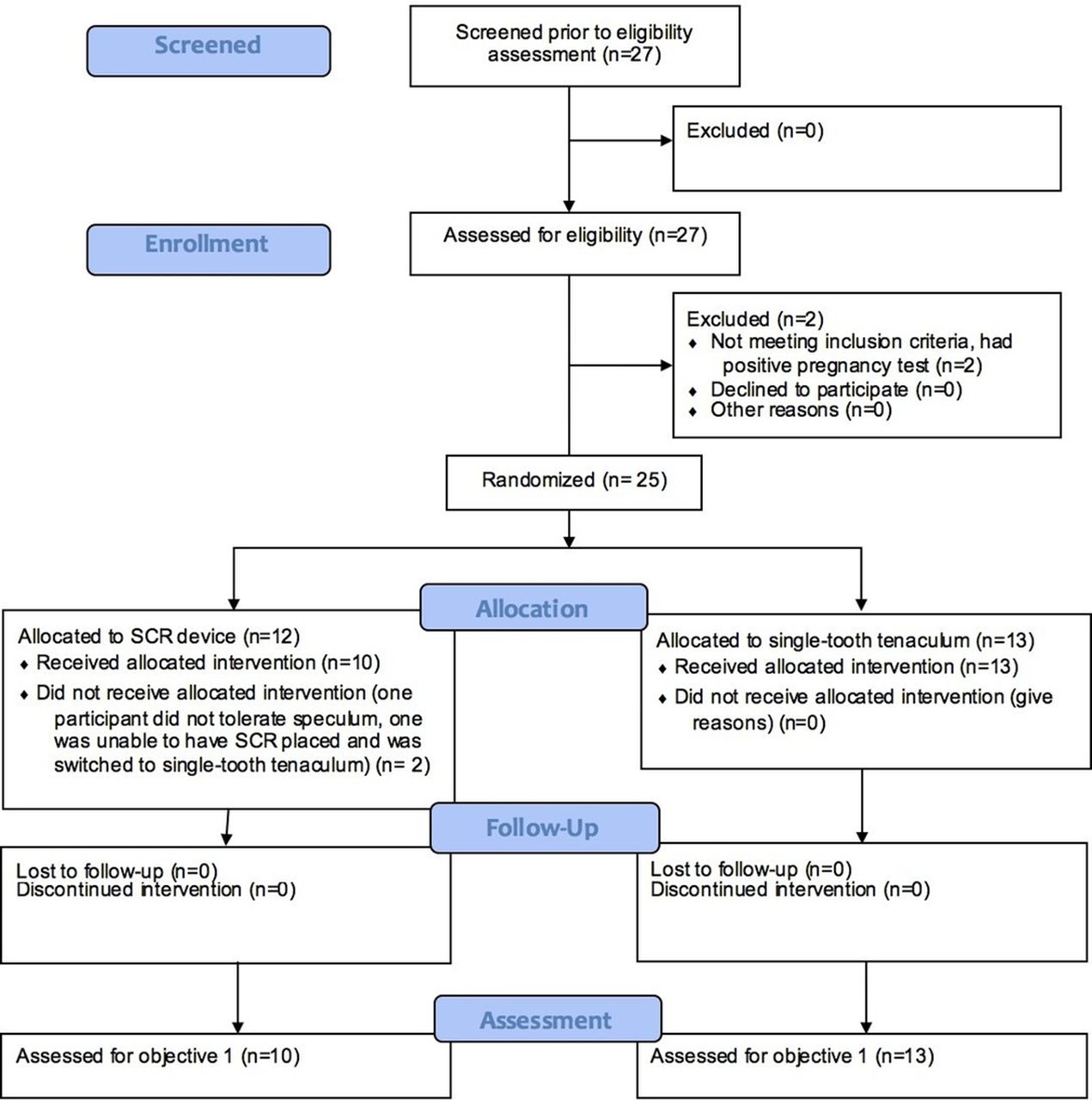
Figure 2 is a participant flow diagram for the Stage 3 randomised control trial (RCT). Most participants chose the levonorgestrel IUD (82% intervention, 77% control). There were no statistical differences in participant characteristics between groups. Of attempted insertions, 96% (23/24) were successful. There were no spontaneous releases of the device from the cervix. No cervical bleeding was noted in any of the intervention procedures and no participants required adjuvant pain medication or local anaesthetic. There was a trend toward lower VAS pain scores for all measurements after baseline for the intervention group, including the primary outome measurements at VAS 3. Almost all (9/10) the women in the SCR group and 100% of the control group reported being ‘satisfied’ or ‘very satisfied’ with the procedure immediately after the insertion. Both groups (30% intervention vs. 31% control) reported some experiences of pain at follow-up; pain scores tended to be lower in the intervention group compared with the control group, though again this was not statistically significant.

{kind=link}
{kind=link}
Bioceptive Pilot Feasibility Study CONSORT flow diagram. SCR, suction cervical retractor.
Adverse effects
One participant (intervention group) withdrew prior to the procedure on account of her inability to tolerate the speculum placement. A second participant assigned to the intervention group did not successfully receive her IUD using the SCR, due to a failure to attach the SCR to the cervix. The participant received her IUD using a single-tooth tenaculum. Her demographic data is included but she was omitted from the subsequent analyses as her VAS scores were not collected per the study protocol. In 3/11 SCR cases, more than one attempt was required to securely attach suction.
Discussion
Main findings
Our current feasibility findings suggest the SCR device has promise in potentially reducing pain associated with cervical traction during the insertion process and is a feasible intervention. Our findings suggest that reported pain scores from participants receiving their IUD using the SCR device trended lower than patients who received their IUD using the standard of care, though these were not statistically significant differences. Thus, these findings cannot exclude the role of chance. The VAS values were within values recently reported for IUD insertion studies.9 18 21 27 Additionally, there were no reports of patient bleeding with the use of the SCR device. Finally, the partial dilation of the cervix with the SCR could potentially make the insertion process easier.
Strengths and limitations
While we strove for generalisability by including nulliparous and parous participants, as well as those with a history of cervical procedures (including loop electrosurgical excision procedure (LEEP)), we were limited by the main study weakness inherent to a feasibility study of this kind - a small sample size. The lower pain scores in Stage 2 relative to Stage 3 are likely due to a sampling aberrancy in the small sample in Stage 2. Our study was not powered to detect statistical significance, but these findings may be helpful for powering future studies testing this hypothesis. Assessing multiple VAS endpoints increases the risk of a type I error. In future studies of the device it will be critical to identify a primary outcome and minimise pain score testing at multiple time points or power the study appropriately for multiple assessments.28 The main study strength is use of randomisation in Stage 3, and use of a validated pain scale in this study provides some ability to validate the values obtained here against other reports.9 27 29
Generalisability and future recommendations
Future research will identify women who are good candidates for the SCR device, as the device may require sizing or specific techniques to accommodate a wide range of anatomic variations. Use of a single, experienced provider to test the SCR device reduced operator-dependent variation; however, assessing provider knowledge, training, acceptability and use should be the subject of future trials on SCR. Finally, there have been studies that have identified reductions in IUD insertion pain through the use of medications.2 6–8 10–13 It is likely that no one technique or device will alleviate all insertion pain; combined studies using both new devices and techniques (such as the SCR) alongside medication protocols should also be considered. These findings suggest the importance of future testing of this device with a larger, more diverse cohort of providers and women. In addition to future research on pain, it will be important for studies to assess other components of SCR use.
Conclusion
This study is the first to report on the feasibility and acceptability of a new, suction-based method of cervical traction. Our findings suggest that the method is sound and warrants future research.
References
Footnotes
Contributors All authors made significant contributions to the research endeavor and manuscript, in accordance with the ICJME criteria for authorship.
Funding This study was funded by Bioceptive, Inc., the company developing the suction cervical retractor. Bioceptive employees were involved in the study design and manuscript writing but were not involved in data collection or analysis. Use of REDCap provided by Eunice Kennedy Shriver National Institute of Child Health and Development grant (8UL1TR000105 (formerly UL1RR025764) NCATS/NIH). Support from the Eunice Kennedy Shriver National Institute of Child Health & Human Development and the Office of Research on Women’s Health of the National Institute of Health is provided under award number K12HD085852 for JNS, K12HD085816 for LMG, and K24HD087436 for DKT.
Competing interests DKT received speaking honoraria from Allergan and Medicines360, served as a consultant for Bioceptive, and served on advisory boards for Actavis, Bayer, Pharmanest and Teva. LMG served on an advisory board for Evofem Biosciences. BC is a biomedical engineer for Bioceptive, Inc. The Department of Obstetrics and Gynecology, University of Utah, receives contraceptive research funding from Bayer, Bioceptive, Medicines360, Merck, Teva and Contramed. The other authors have no conflicts of interest to disclose.
Patient consent Obtained.
Ethics approval University of Utah IRB.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Interested parties may contact the lead author for a copy of a de-identified dataset should they be interested.
Linked Articles
- Highlights from this issue